- 脑胶质母细胞瘤血管形态和模拟
- 发布于 2015-10-16 23:55 来源:顾建文医生
Glioblastoma vasculature and vascular mimicry
Introduction
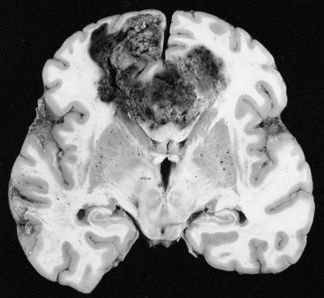
Glioblastoma (GB) is the most common intracranial malignancy with an annual incidence of approximately 3 C 5 cases per 100,000 individuals. In terms of overall cancer burden GB is uncommon, however, the combination of brain localization, invasiveness, and extremely poor prognosis make it one of the most feared of all cancer diagnosis. Glioblastoma currently has an overall median survival time of 12 C 15 months despite maximally combined therapy of neurosurgery, radiation, and combination thermotherapy. It is interesting to note that the extremely slow development of effective therapy/treatment lies in stark contrast to the rapidly expanding foundation of knowledge regarding the molecular pathogenesis of this disease. The vast majority of research in the field of neuro-oncology has been published in the last 20-30 years, largely driven by a greater understanding of brain tumor genetics. More recent molecular classification has provided the basis for the existence of four distinct types of high-grade primary glioblastoma: classical, mesenchymal, neural, and pro-neural subtypes. These subtypes are differentiated molecularly based upon various proteomic features, signature mutations, methylation features, and currently identified tranional regulators. The comprehensive molecular classification of high-grade gliomas is just now starting to transform current classification, which currently follows the consensus WHO histopathological criteria. Despite sophisticated molecular classification schemes, a relatively high percentage of gliomas remain difficult to reproducibly categorize due to considerable histological overlap. Phenotypically, the tumor displays marked hypercellularity, serpiginous areas of necrosis, and expansive endothelial cell proliferation with tumor vasculature being torturous, disorganized, and highly permeable. Grade IV neoplasms exhibit extremely aggressive proliferation of endothelial cells as compared to Grade II and Grade III tumors. The increased vascular permeability compounds the issue by leading to increased cerebral edema and inflammation. Abnormalities in the endothelial walls, pericyte coverage, and basement membrane also result in loss of structure and function of the critical blood brain barrier.
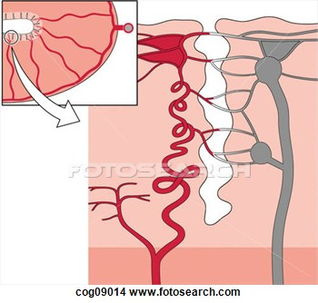
Glioma vasculature formation occurs through at least three distinct processes. 1) Angiogenesis; the process of generating new blood vessels from rerouting and remodeling of pre-existing vessels, 2) Vasculogenesis; classically considered an embryonic process, but has since been identified in tumors as the de novoformation of primitive blood vessels by the differentiation of circulating bone marrow-derived endothelial progenitor cells, and 3) Vascular Mimicry (VM); recently identified to provide a contribution to tumor vasculature by trans-differentiation of glioma cells into tumor-derived endothelial cells (TDECs).
Molecular pathways
It is said that cancer metastases, regardless of the primary tumor, play by a different set of rules from the primary tumor, and cancer deaths primarily result from invasion and metastases that are resistant to conventional therapies that may otherwise effectively treat the primary tumor. Tumor survival is dependent upon a rich blood supply needed to sustain tumor growth and further metastasis. Malignant gliomas, like other neoplasms, require angiogenesis to establish a source of nutrients/oxygen and to eliminate cellular waste products. The tumor vasculature also creates a localized “niche” microvascular environment within which the tumor-initiating cells may be able to effectively resist therapy. Angiogenesis is a key pathologic event and necessary for the progression of a solitary localized neoplasm to a highly aggressive tumor. This critical tenant subsequently ignited the field of neoplastic angiogenesis research, which has focused on targeting endothelial cells forming the neovasculature of growing tumors and served as the major organizing principle for drug development of various clinical trials. Because GB is one of the most vascular rich tumors and VEGF (vascular endothelial growth factor) is produced by tumor cells, anti-VEGF antibody (bevacizumab/Avastin) has been used in clinical trials. The results of clinical trials are disappointing to say the least, more than half of patients with GB only transiently respond to combination treatment of Avastin and Irinotecan. Mechanisms proposed ot explain resistance to anti-VEGF therapy include 1) Activation of other pro-angiogenic signaling pathways 2) Recruitment of bone marrow (BM)-derived myeloid cells that protect and nurture vascular cells and/or 3) Protection of blood vessels by increased pericyte coverage. In GBs, the antitumor effect of the antiangiogenic therapies is likely due to normalization of vasculature, which also decreases edema. The disappointing results of the angiogenesis inhibitor trials, together with new evidence generated from molecular and animal research on human GB tumor progression, have given insights into the molecular mechanisms underlying the perfusion of tumors, particularly those expressing an aggressive invasive phenotypes such as GB.
In tumor angiogenesis, bone marrow-derived endothelial cell precursors (ECPs) have historically been known to be the main source of the vascular endothelial cells. However, recently it was shown that BM-derived endothelial precursor cells did not solely contribute to the vascular endothelium, and it is now understood that TDECs in GB transdifferentiated from neuroectoderm and that tumor cells themselves can be involved in tumor angiogenesis. This more recent evidence suggests an alternative mechanism, one whereby tumor microvasculature is derived directly from tumor cells, and this process is called Vascular mimicry (VM). VM describes the functional plasticity of aggressive cancer cells forming de novovascular networks, providing perfusion pathways for expanding tumors, transporting fluid from leaky vessels, and/or connecting to endothelial-lined vasculature. VM evidence suggests that this blood-perfused microvasculature plays a critical role in tumor development and is independent of endothelial cell angiogenesis. Interestingly, VM networks have also been shown to exhibit anticoagulant properties through local expression of anticoagulant molecules with the overall goal to facilitate the flow of blood into and among aggressive tumors. The original study describing VM was based on evidence that showed transport of injected fluorescent dye throughout VM networks. Subsequent experimental evidence has shown real-time in vivophysiologic perfusion of blood between endothelial-lined mouse vasculature and VM networks in human tumor xenografts using Doppler imaging of microbead circulation. This study went on to show that aggressive melanoma cells are capable of de novo three-dimensional vascular structure formation, this finding was subsequently validated by high resolution Electron Microscopy showing the morphological/structural details of the tumor-formed vessels and similarities in ultrastructure between VM and traditional endothelial-lined vasculature. Melanoma studies also showed tumor cells may co-express endothelial, embryonic/stem cell, and tumor cell markers. A really interesting study that describes hypoxia as a catalyst of VM phenotype was demonstrated with the transplanatation of human metastatic melanoma cells into an ischemic mouse limb, which resulted in the formation of a blended vasculature composed of human melanoma and mouse endothelial cells. After blood flow to the limb was sectored, the melanoma cells formed a large tumor, this study demonstrates the amazing influence of the microenvironment on the transendothelial differentiation of melanoma cells, which reverted to a more tumorigenic phenotype as the environmental cues changed. Transdifferentiation of GB stem cells (GSCs) into mural-like (vascular smooth muscle/pericyte-like) tumor cells highlights the plasticity of GSCs. The plasticity of GSCs is further displayed by the multi-potency of neuronal stem cells that are capable of differentiating into several cell lineages (i.e., astrocytes, neurons, and oligodendrocytes); and into a variety of cells such as blood cells, muscle cells, and vascular endothelial cells.
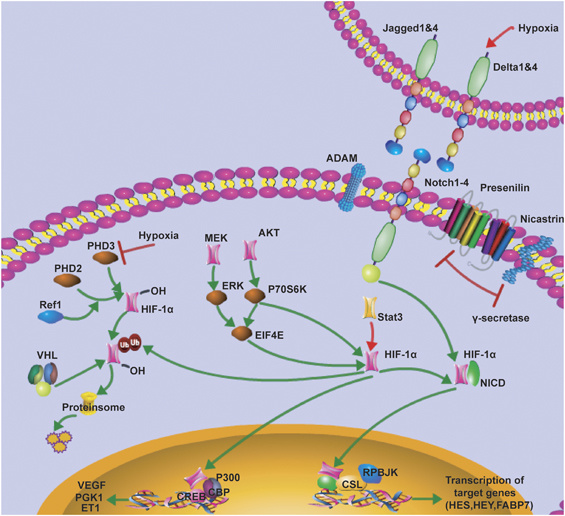
Many molecular details underlying VM have been deciphered, but as with any other topic of scientific research, there are still many more questions than answers. However, it is known that critical VM-modulating genes are associated with vascular (VE-cadherin, EphA2, VEGF1) embryonic and /or stem cells (Nodal, Notch4), and hypoxia related (HIF, Twist1) signaling pathways. A few selected common genes and pathways will be discussed briefly in this article. 1) Notch and Nodal signaling pathwaysare tow pathways that have been shown to be critical both for embryonic stem cell regulation and tumor cell behavior; interestingly, crosstalk between these pathways regulates tumor cell behavior, aggressiveness, and VM network formation. Nodal signaling modulates vertebrate embryogenesis functioning in left-right asymmetry determination and stem cell pluripotency; generally absent in adult tissues but is known to become reactivated in aggressive cancer. Noteworthy is the fact that aggressive cancers reactivate Nodal, but not its regulatory protein Lefty, therefore Nodal signaling proceeds unchecked and is able to promote aggressive tumor cell behavior. Notch signaling is important in stem cell differentiation and self-renewal and is expressed in various embryonic and adult tissues. 2) Hypoxia-Inducible-Factor (HIF) complex is a key regulator of oxygen homeostasis in both physiological and pathological environments. HIF can modulate and cross talk with both Notch and Nodal signaling pathways. HIF over-expression in cancer induces the expression of gene products involved in angiogenesis (i.e., VEGF). It is important to note that hypoxia has been shown to induce both Notch and Nodal pathways via HIF1-alpha/2-alpha and HRE signaling. There is significant cross-talk between HIF1-alpha and Notch-signaling, overall working within tumor cells to promote an undifferentiated cellular state. This is how it becomes conceivable that therapeutic usage of antiangiogenic agents may counterintuitively promote tumor plasticity and metastatic/invasive progression.
Genetic analysis of individual ECs via microdissection followed by genetic analysis of GB ECs showed that 50-90%; of ECs in the GB tumors carry the genetic abnormalities found in the tumor cells themselves, suggesting a common origin. Furthermore, mouse experimentation has revealed that the tumor-derived endothelial cells originated form tumor-initiating cells and did not result from the cell fusion of ECs and tumor cells. An in vitrodifferentiation assay in the same study suggested that hypoxia was the critically important factor in the differentiation of GB tumor cells to ECs and is independent of VEGF. Tumor derived endothelial cell (TDEC) formation was not only resistant to anti-VEGF receptor inhibitor, but it led to an increase in their frequency. It can be surmised that that TDECs make an important contribution to the resistance of GB to anti-VEGF therapy, and it can therefore also be a potential target for GB therapy. By and large, tumor cell VM nicely illustrates the functional plasticity of the aggressive cancer phenotypes and serves as a selective advantage for rapidly growing tumors in need of perfusion. VM can provide one of several sources for a tumor blood supply that can directly or indirectly interact with other vasculature. The literature is in agreement what the underlying induction of VM seems to be related to hypoxia, which in turn activates a variety of genetic alterations as previously described that then directly promote the transendothelial phenotype of tumor cells capable of VM.
Treatment Obstacles
As previously mentioned, the detailed and precise deion of the molecular biology of glioblastoma lies in stark contrast to the lack of advances in therapy. With a currently expanding knowledge of molecular biology, more potential targets have been identified and the key will be to develop potent inhibitors and effectively use combinations of them in an appropriate and targeted manner. A major limitation to targeted drug delivery is that migrating primary tumor GB cells blend in with normal tissue, are difficult to identify and target, and do not elicit an angiogenesis response. Additional therapeutic obstacles include 1) No effective agonists/antagonists against identified targets, 2) Therapeutic molecules must pass BBB in order to reach invading cells, including those located at a distance from the central part of tumor. Varying degrees of BBB dysregulation exist within a given GB tumor, by itself, the BBB is known to present a challenge to effective transit of therapeutic molecules to the brain, and continued study of BBB in tumor setting s is needed to lend insight into effective drug delivery in GB, and 3) currently lack ideal animal model for GB treatment studies.
Combined antivascular therapy aiming at both mural-like tumor cells and endothelial cells, in addition to cytotoxic drugs targeting tumor cells may hold promise. It seems plausible that the most efficient way to target tumor cell plasticity is to inhibit multiple signaling pathways simultaneously. The molecular pathways that have been experimentally identified as critically involved in VM. The molecular pathways that have been experimentally identified as critically involved in VM, along with failed clinical trial data should serve as a strategic roadmap for drug development with goal being to overcome tumor cell plasticity, drug resistance, neoplastic angiogenesis, invasion and metastasis. The emerging data on embryonic pathways, Notch and Nodal, which are reactivated in aggressive tumor cells, may provide valuable therapeutic targets that exploit the convergence of embryonic and tumorigenic signaling. Suppression of these pathways results in the inhibition of VM, tumorigenicity, and the reversion of the stem cell-like phenotype to that of a differentiated cell type. Simultaneous targeting of the Notch and VEGF pathways may perhaps provide a more viable combination approach to target cancer stem cells with anti-VEGF therapy.
Conclusion and Future Work
Several centuries of research have shown the high degree of plasticity associated with aggressive neoplasms. With the recent addition of sophisticated molecular tools, the exact mechanisms, etiology, and implications of tumor cell pathobiology have been further elucidated. During this time, research has demonstrated that the presence of a small population of GSCs is closely associated with resistance to radiotherapy and anti-angiogenic therapy. However, clinical treatment using an anti-VEGF therapy alone has routinely failed to significantly affect patient survival and clinical outcomes. It is logical to speculate that as part of its ability to survive in the presence of anti-angiogenic agents and reemerge from dormant primary tumors, GSCs may undergo an alternative vascularization that develops mural-like cell-associated networks and nourishes the bulk of the growing tumor cells. From the literature within the field of neoplastic angiogenesis, it is now appreciated that the tumor vasculature is highly complex and can be derived from a variety of sources, including angiogenic vessels, cooption of preexisting vessels, mosaic vessels lined by both tumor cells and endothelium, and postnatal vasculogenesis. Furthermore, recent studies have shown the tumor origin of endothelial-like cells, vascular mimicry, in specific cancers, further complicating the strategies for targeting a genetically unstable and heterogeneous vasculature.
The commitment to genomic characterization of GB has fueled substantial progress in understanding of this cancer, particularly within the past 5-10 years. Extensive genomic characterization has provided a high-resolution image of the various molecular alterations underlying GB, and suggests that indeed this disease represents several histologically similar, yet molecularly heterogeneous diseases. The heterogeneous nature of this neoplasm, both within and across tumors, underscores the difficulty in developing efficacious treatment and provides a challenge both to annotate tumors and to stratify patients for trials and treatment. Despite tremendous progress in understanding of the genetic basis of glioblastoma, targeted therapeutic approaches based on known genomic alterations have yet to be proven efficacious. The morphological heterogeneity that prompted the original deion of high-grade glioma as, “multiforme”, has indeed extended to the molecular level. It is likely that intratumoral heterogeneity and target cooperativity conspire to create a multiple dependency state whereby single-target inhibitors are not sufficient to significantly attenuate tumor growth. There is also an incomplete understanding of the functional consequences of many of the mutated genes in GB. Research is just now beginning to understand how this molecular heterogeneity is manifested and what exactly the functional consequences of these genetic alterations are. It is also not clear if these intermingled cell populations with unique genomic alterations behave as independent tumors or are interdependent with each other. In addition to understanding the molecular basis of GB heterogeneity, it will also be important to consider the contributions and phenotypes of the diverse non-tumor cell types, like stromal and inflammatory cells that also populate the glioma microenvironment. Further work is indeed necessary to fully understand GB biology and will require integrated studies, including genomic, animal models of disease, and as always a careful study of human tissue.
