- 医学科普:黏多糖贮积症都有哪些临床表现
- 发布于 2014-11-09 19:06 来源:刘舒医生
黏多糖贮积症(mucopolysaccharidosis, MPS) 是一组由于溶酶体酶缺陷造成的酸性黏多糖分子不能降解而在溶酶体内累积的疾病,该病主要以骨骼畸形、身材发育迟缓为主要临床表现,伴或不伴智力发育迟缓、心脏瓣膜病、角膜混浊和耳聋等。
根据临床表现和酶缺陷,粘多病可分为Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅳ、Ⅵ、Ⅶ、Ⅸ等8型(无Ⅴ及Ⅷ型)。各型MPS由于涉及组织及程度的不同而导致临床表现和后续的治疗、预后有所不同,但各型MPS的临床表现,有其共同性和一致性。为了方便广大家长进行初步临床鉴别,内容见下:
以经典的Ⅰ型患者为例,主要有以下临床特征:
1、面容粗陋:头大,前额突出,眉毛浓密,眼球突出,眼睑肿胀,鼻梁低平,鼻孔上翻。嘴唇大而厚,舌大,牙龈增生,牙齿细小且间距宽。 皮肤毛发浓密粗糙,发际线低。
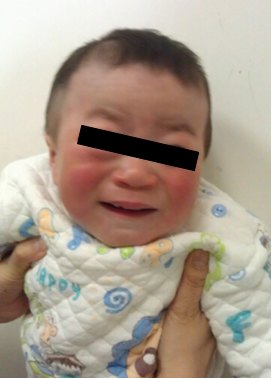
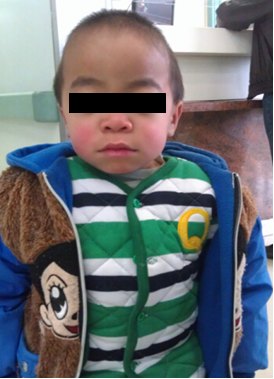
2、角膜混浊:随着病情的进展,角膜混浊逐渐加重,最后可导致失明。
3、肝脾肿大:腹部膨隆,腹腔压力大导致脐疝和腹股沟疝频发。
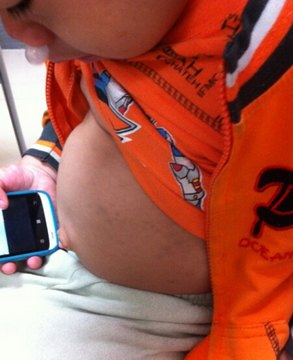
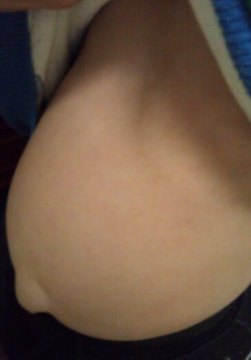
4、身材矮小:患儿脊柱后凸,体格生长发育迟缓,伴脊柱、胸骨、肋骨发育畸形。

5、关节僵硬:累及各关节,使这些关节的活动度受限,关节挛缩,如指关节受累,显示出“爪形手”的特征;如累及膝关节和髋关节等大关节,则导致步态不稳,走路易摔跤。

6、智力落后:患者在1岁左右可能就表现有智力落后或者倒退,甚至出现严重智力障碍。
7、听力障碍:听力障碍主要是由于中耳小骨发育障碍所导致的咽鼓管功能紊乱和反复中耳感染、鼓膜瘢痕、第八对颅神经损伤所致。
8、心脏:大部分患者的心脏累及在疾病的后期,表现为瓣膜病,可导致心力衰竭。
其他类型的MPS:
Ⅱ型MPS患者症状较Ⅰ型稍轻,因该型是X连锁隐性遗传,故患儿均为男性,患者的角膜也不浑浊;Ⅲ型MPS患者以严重智力落后为主要的临床表现;Ⅳ型MPS患者骨骼畸形较为严重,如腕关节松弛,胸廓向前突出形成类似鸡胸,同时改型患儿有角膜混浊,但智力一般正常;Ⅵ型患者智力正常,但角膜混浊明显;Ⅶ型患者临床表现差异可非常大,严重的表现为胎儿水肿,轻型的患者可只有身材矮小和骨骼畸形等。
