- 高血压、周期性血尿
- 发布于 2009-08-30 14:59 来源:孙首悦医生
患者,1976年11月出生,33岁,社会性别男性,因“血压升高17年伴反复乏力10年,周期性血尿半年”入院。患者于17年前体检时发现血压升高,约140/100mmHg,当时无头晕、头痛、胸闷、心悸等不适,未重视。10余年前开始无诱因下出现全身乏力,走路跌倒,于当地医院就诊,查血钾偏低,给予口服补钾,1周后症状好转,以后类似症状多次出现,自服氯化钾后自行缓解。5年前因测血压仍高,最高达210/110mmHg,开始不规则服用一些降压药物。9个月前患者在夜晚安静状态下突感右侧肢体无力,活动困难,持续30分钟后自行好转,未予特别重视。7个月前再次在相同情形下突发头晕、右侧肢体无力,较前一次加重,右上肢完全不能活动,手指屈曲不能伸直;右下肢肌力减退,行走需搀扶。在当地医院测血压174/129mmHg,MRI示“左脑室体旁梗死灶,双侧额顶叶多发缺血灶,左外侧裂区蛛网膜囊肿引起”,测血钾2.47mmol/L,血皮质醇(F)、促黄体素(LH)、促卵泡素(FSH)降低,孕酮(P):11.9 ng/ml、睾酮(T):6.7 ng/ml均升高,雌二醇(E2) 、醛固酮正常。肾上腺CT示“右肾上腺明显增粗,左肾上腺占位”。进一步检查发现,患者的外生殖器表观为男性,但阴囊中无睾丸,阴茎短小,有尿道下裂,染色体分析提示为46XX,99锝扫描未见睾丸显像,B超检查在患者盆腔内发现女性内生殖器,但子宫小、双侧卵巢多发小囊肿。诊断为先天性肾上腺皮质增生症(CAH),继发性高血压。给予治疗方案为醋酸可的松早8时25mg,午2Pm 12.5mg,安体舒通60mg/日、依那普利10mg/日、拜心同30 mg/日。用药后,有双侧乳房隆起伴感胀痛,2个月前出现每月一次的规律性血尿,无尿频、尿急、尿痛、排尿困难,持续4-5天自行消失。
追问病史,患者父母非近亲结婚,患者为足月、顺产,出身时体重、身高正常,但外生殖器性别认定模糊,“阴茎”短小伴“龟头”粘膜潮红湿润。9岁前生长发育过快,身高超过正常同龄儿童,9岁时身高即达137cm,但此后未再继续长高,身高逐渐落后于同龄人。并出现全身毛发明显增多,以四肢较明显,伴有皮肤黏膜色素沉着。成年后患者与女性交往并订婚,有性行为但因“阴茎”勃起后短小而性生活质量不高。患者智力正常(学习成绩中上),幼年时期易“感冒“。母亲曾在患者之前怀两胎,但出生后都夭折。现有一弟弟,身体健康。
体格检查:身高:137cm,体重:88kg,BMI:22.9Kg/m2,上部量:77cm,下部量:60cm,臂展:131.5cm。神智清楚,精神好,言语流利,右足跛行。阴毛腋毛浓密,Ferriman-Gallway毛发评分27分,乳房发育TannerⅡ期。心率60次/分,律齐,心音有力,心尖部可闻及Ⅱ-Ⅲ级杂音。右上肢体肌张力增高,呈铅管样,肌力Ⅳ级,左侧肌张力,肌力正常,右侧手指呈蜷缩样,无法自行伸直,双侧肱二头肌/骨膜反射(++),Hoffmann征(+),右下肢肌张力增高,膝反射(++++),踝反射(++++),膑阵挛(+),踝阵挛(+),肌力正常,左下肢肌力,肌张力正常,膝反射(++),右侧巴氏征可疑阳性,左侧巴氏征(+),两侧Gordon征、Oppenheim征、Chadock征、Romberg(-)。双侧足背动脉搏动正常,生理反射存在,病理反射未引出。外阴部男性外观,阴茎短小,尿道开口于阴茎根部,阴囊内睾丸缺如。
辅助检查:肝肾功能、血电解质、血脂、生长激素、甲状腺功能、皮质醇、血浆肾素活性-血管紧张素II-醛固酮、OGTT+胰岛素释放正常,促肾上腺皮质激素(ACTH) 539.80 (正常值:12.0~78.0 pg/ml),性激素:LH 3.79 (卵泡期正常值:2.6~26.5 mIU/ml),FSH 4.19 (卵泡期正常值:3.4~21.6 mIU/ml),E2 25.00 (卵泡期正常值:35~169 pg/ml),P 2.20 (卵泡期正常值:<0.1~0.3 ng/ml),催乳素(PRL) 28.81 (正常值:1.2~29.9 ng/ml),T 0.19 (正常值:0.1-0.8ng/ml),17-OHP 14.96 (0.40-1.02 ng/ml),性激素结合球蛋白(SHBG) 22.60 (19.8~155.2nmol/L),DHEA-S 8.70 (95.8~511.7μg/dl)。妇科B超:盆腔内见女性生殖器官,子宫内膜厚5mm,左卵巢内见卵泡样无回声区8枚,单切面最大直径13mm,右卵巢内见卵泡样无回声区10枚,直径5-8mm。乳腺B超:男性乳腺发育。垂体MR平扫:垂体形态饱满,双侧基底节区多发异常信号灶。肾上腺CT:双侧肾上腺先天性增生伴左侧肾上腺髓样脂肪瘤,右侧肾上腺腺瘤。
放射科方文强主任读片:
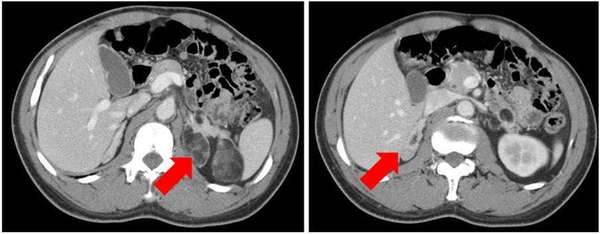
头颅MR平扫+弥散成像读片:双侧基底节区见小片T1W呈低、FLAIR呈低信号影,双侧基底节区、额叶、顶叶见散在小斑片状异常信号灶,T1W不明显呈等和稍低信号,FLAIR病灶信号未被抑制呈高信号,DWI病灶信号未见明显增高。各脑室、脑池及脑沟未见明显增宽、扩大或变窄。中线结构无移位。所示诸副鼻窦区未见明显异常信号。影像学诊断为:双侧基底节区脑梗塞,双侧基底节区、额叶、顶叶散在小缺血灶。肾上腺CT读片:可见左侧肾上腺增粗,并可见不规则混杂密度影,内可见负性脂肪密度影,软组织密度影及斑点状钙化灶,增强扫描实性成分有所强化,右侧肾上腺结节状增粗,右肾上腺外侧支类圆形密度灶,直径约为1.09cm,增强扫描强化较明显。影像学诊断为:两侧肾上腺先天性增生伴左侧肾上腺髓样脂肪瘤,右侧肾上腺腺瘤可能。结合患者的临床表现和实验室检查,先天性肾上腺皮质增生症诊断明确,其肾上腺的影像学表现,不仅有整个肾上腺皮质的增粗,还可以看到肾上腺髓样脂肪组织的增生,目前认为,这与肾上腺网状内皮细胞或肾上腺内质细胞在长期异常升高的ACTH等因素刺激下产生化生,致使髓样脂肪组织发生改变所致。若要进一步确诊,可以行手术或穿刺活检。
孙首悦主治医师:
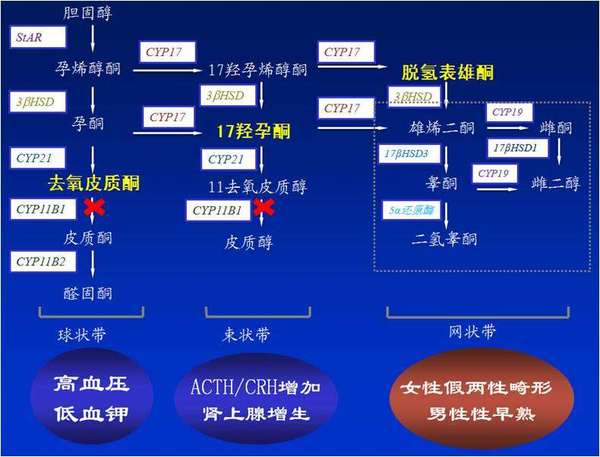
综合患者的病史、临床表现及各项辅助检查,有如下特点:社会性别男性,而染色体性别为女性,为女性假两性畸形患者。出生时性腺性别模糊,有女性内生殖器官而外生殖器更趋向男性而被当作男孩抚养,幼年时生长过快,男性性征很早出现且异常明显,但在10岁前就已停止身高增长,最终身高矮于正常人,提示患者出生前即有高雄激素血症并始终伴随其生长发育过程。另一方面,患者有长期的青少年时即已起病的高血压,早已对其耐受而延误治疗,以致在40岁前就出现卒中,同时还伴有低钾血症,但醛固酮水平正常。相对于过度升高的ACTH,血、尿F水平只在正常范围,提示肾上腺皮质功能相对不足,同时,我们还观察到17-OHP的升高。影像学上有肾上腺皮质的增生,先天性肾上腺皮质增生症(CAH)的诊断明确,综合参考各项指标,考虑为11β羟化酶缺陷症型,这一点从基因诊断得到进一步证实。当地医院的内分泌医师也考虑到了这一罕见的疾病而给予糖皮质激素替代,和降压药控制血压治疗,同时考虑到患者有低钾血症和高雄激素血症并存而选用了较大剂量的安体舒通来治疗,以期在拮抗盐皮质激素降压升血钾的同时可以拮抗高雄激素的作用。治疗显然有效,患者血压下降,血钾恢复正常并有乳房增大伴有胀痛,而且在雄激素水平下降后,对垂体促性腺激素的抑制解除,卵巢功能也得到恢复,患者甚至出现了子宫内膜的增殖和周期性脱落,在30岁时有了月经初潮,由于外生殖器的畸形,阴道的前三分之一与尿道融合有共同开口,因而经血排出体外时犹如“血尿”持续数天,在糖皮质激素替代下,月经周期规律,从而形成了周期性“血尿”。目前存在的问题是尽管有糖皮质激素-可的松的替代,患者的ACTH仍远高于正常,患者的社会性别是男性,心理性别也是男性,对于突如其来变化在心理上还没有准备好如何去面对,无法立刻接受性别的转变。因此,有必要调整用药来解决问题,并有必要多给患者一些时间作为心理缓冲期逐渐接受适应变化。
王卫庆教授:
对于CAH,11-β羟化酶缺陷症,应与原发性醛固酮增多症、嗜铬细胞瘤、库欣综合征、肾上腺肿瘤、CAH的其它类型如21-α羟化酶缺陷,17-α羟化酶缺陷或3-β羟类固醇脱氢酶肾上腺皮质功能减退、肾上腺结核、肾上腺淋巴瘤等疾病作鉴别。因为CAH的皮质醇缺乏只是相对不足,故治疗上应选用中长效的糖皮质激素如地塞米松用作补充治疗来抑制过高的ACTH分泌。对于未成年的儿童CAH患者,地塞米松的应用对骨骼生长发育的抑制作用较强,故选用分子结构与皮质醇更为相似的短效的可的松来治疗为宜,但对ACTH的抑制作用不如地塞米松持久而稳定。该患者9岁以后不再长高,骨骺已闭合,提示骨骼已定型,不存在糖皮质激素对骨骼发育的影响,因此可以选用长效糖皮质激素地塞米松,以0.75mg作为起始剂量,每晚睡前服用。由于11-β羟化酶的缺陷,醛固酮的合成不足,其前体物质脱氧皮质酮(DOC)大量堆积,而DOC有强大的理盐激素和理糖激素作用,可以代替醛固酮,DOC过量也可造成高血压、低血钾。从发病机制上讲,只要适当服用糖皮质激素补充治疗,抑制ATCH过多分泌对肾上腺皮质的刺激,DOC的产量自然会减少,从根本上解决问题,而安体舒通等只是在受体层面减少激素的作用,并不减少DOC的产量,作为首选用药欠妥,安体舒通还有拮抗雄激素的作用,患者的使用剂量较大,出现了乳房发育的迹象,对于女性来说,这或许是件好事,但对于患者来说,还不能完全接受性别的改变,或者说,患者还没有决定将来性别的取向,故建议停用安体舒通。在降压药的选择上,首选ACEI和CCB类药物控制血压,并联合抗血小板聚集的药物。
泌尿外科孙福康主任:
患者诊断CAH明确,在一些CAH的患者的肾上腺影像学上可以发现肾上腺髓样脂肪瘤的表现,这个患者也有如此表现。肾上腺髓样脂肪瘤有时与肾血管平滑肌脂肪瘤在影像学上难以区别,要依靠病理诊断。无论如何,这二者都是良性肿瘤,且预后良好,但如果肿瘤短时间内快速增长或出血也可能危及生命。原则上大于3.5cm的瘤可以考虑手术切除,由于患者近期发生过卒中,目前无手术指征,建议患者继续内科治疗,定期随访肾上腺CT,必要时考虑手术切除髓样脂肪瘤,病理检查明确诊断。
宁光教授:
CAH的发病率约为1:10000,21-α羟化酶缺陷最为常见,而11-β羟化酶缺陷国内少见报道,目前在本科已诊断4例。由于该病常以高血压伴肾上腺增生为首发症状前去就诊,极易被误诊为肾上腺肿瘤而经历一侧肾上腺的切除术,而另一侧的肾上腺增生更为显着,术后患者的症状将更加严重。因此,对于年轻起病的高血压伴有低血钾,男性有性早熟,女性有假两性畸形的表现,同时有双侧肾上腺增生的患者,均应该想到有罹患该病的可能,应予以ACTH、皮质醇节律、17-OHP、性激素、肾素-血管紧张素-醛固酮等方面仔细筛查予以排除。该患者症状典型,诊断明确,如此女性男性化,周期性“血尿”表现实属罕见。目前的治疗应以尽快解除高血压、低血钾对机体的损害为主,不宜盲目手术。在患者今后的性别取向上也不宜匆忙决定,应给患者充分的时间考虑,必要时请心理科医师进行心理疏导,充分尊重患者的想法,制定有效的治疗方案。
患者随访:
根据我科制定的治疗方案,经过5个月的治疗后,多毛,肤黑等症状明显缓解,无低血钾发作,乳房胀痛缓解,每月一次周期性“血尿”规律,但近二次出现“血尿”期间伴有尿频,尿常规中发现白细胞(+),给予抗感染治疗后好转。血电解质正常,ACTH 31.1 pg/ml,P 0.60 ng/ml,T 0.30 ng/ml,17-OHP 3.19 ng/mljun均有所下降。LH 10.17 mIU/ml,FSH 3.78 mIU/ml,E2 39.00 pg/ml,妇科超声提示多囊卵巢。服药后时感中上腹轻度不适伴返酸,加用潘托洛克作为胃黏膜保护剂。征询患者意见,认为暂不做性别取向,保持现有状态,继续维持原治疗方案,定期复查各项内分泌指标。
图一:11β羟化酶缺陷的病理生理过程示意图
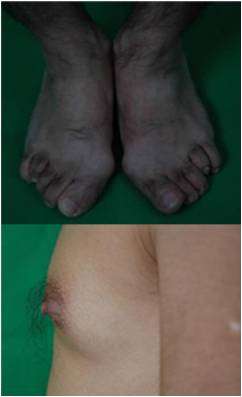
图二:身材矮小,四肢多毛,手足小,皮肤关节皱褶出色素沉着,乳房Tanner II期
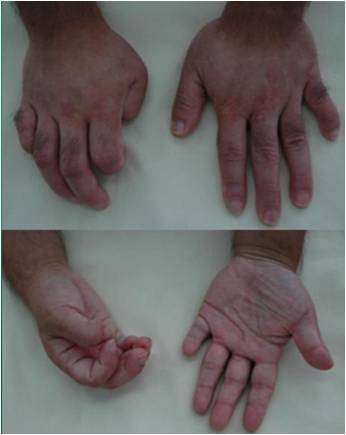
图三:阴蒂如小阴茎,大阴唇融合如阴囊,睾丸缺如,尿道下裂

图四:双侧肾上腺增生伴左侧髓样脂肪瘤,右侧结节