- 杜氏肌营养不良(转载)
- 发布于 2016-02-26 10:24 来源:任淑敏医生
-
假肥大性肌营养不良(DMD)
 郑州大学一附院遗传与产前诊断中心任淑敏
郑州大学一附院遗传与产前诊断中心任淑敏
假肥大性肌营养不良, 也称 Duchenne型肌营养不良症(DMD) ( OMIM 310200 ) 。是最常见的一类进行性肌营养不良症。患病率为 3.3/10万,占出生男婴的20-30/10万,为X-连锁隐性遗传。主要是男孩发病,女性为致病基因的携带者。通常5岁左右发病。肌萎缩是进行性的,这是本病的特点,预后差。本病的发生是由于编码dystrophin的DMD基因的突变所引起, 有约 1/3 病例为散发,没有家族史,是由基因新突变造成。 Becker 肌营养不良 (Becker muscular dystrophy, BMD) 的产生也是由于 DMD 基因突变所引起,通常突变后产生的异常 DMD 蛋白仍具有一定功能,因而临床症状较 DMD 轻得多。
【临床表现】
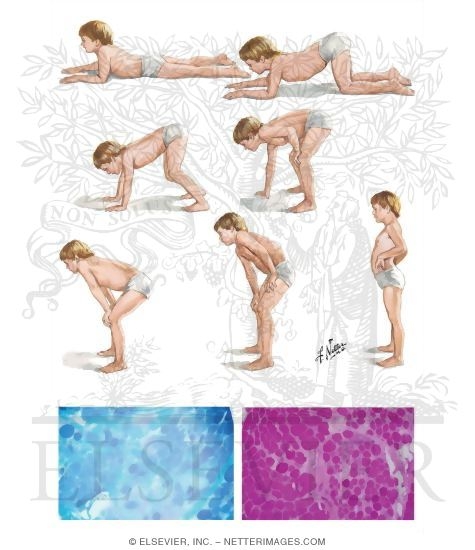
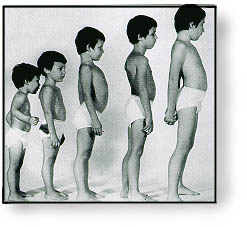
患儿出生时的活动如抬头、坐姿等均正常,自 1 岁以后开始逐渐出现站立和行走困难,首先影响骨盆带肌肉,以后累及肩胛带肌肉。患儿动作笨拙,易跌倒,走路摇摇晃晃,登楼梯或由坐、卧位起立困难。随着病变的进展,臀中肌无力导致行走时呈特殊的鸭步,患儿从仰卧位起立时需先翻转为俯卧,再以双手支持地面和下肢缓慢地站立,称为 Gower 氏征。患儿双侧腓肠肌逐渐呈假性肥大,腱反射减弱或消失。部分患者表现为行为异常。病变呈进行性加重,常到 10 岁时已不能行走,大多数患儿最终卧床不起,并发痉挛、褥疮、肺炎而在 20 岁前死亡。 BMD 患者的临床症状一般较轻,可存活到成年以后。
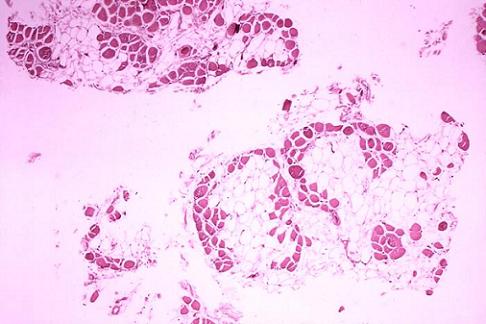
约 1/4 的患儿有智力低下,血中磷酸肌酸激酶( CRK )浓度显著升高,可达正常值的 1万倍,肌电图显示肌病的改变伴有较轻的失神经支配电位;肌肉活体组织检查可见肌纤维坏死与再生同时存在,并有结缔组织增生。
【诊断与防治】
根据患者特有的症状和体征,结合血 CPK 酶学检查和肌电图检查结果,一般不难作出诊断。确诊可根据多重 PCR 技术、 Southern 杂交、点突变检查等方法。对于突变未明确的家系,可用 STR 位点进行连锁分析,用于携带者的检出或产前诊断。
目前对于 DMD 尚无有效疗法,唯一有效的预防途径是对高风险胎儿进行 产前诊断 ,确诊后流产。
【遗传咨询】
本病属 X 连锁隐性遗传病。致死性散发性 X -连锁隐性遗传的 1/3 病例都是由新发生的基因突变引起 ( 又称 Haldane 规律 ) ,这在家系分析时应特别注意。
常见咨询问题:
1.家族中没有发现类似病例,是否也有可能患 DMD?
前面已经提到, DMD中,有约1/3病例是由新发生的基因突变引起,即父母双亲中,DMD基因都正常,但个体胚胎发育时,出现了DMD基因的突变,导致疾病的发生。这种突变发生的个体,可将致病基因遗传给后代。
2.如何通过性别选择,规避 DMD的遗传风险?
本病主要为男性发病,在已生育过 DMD患者的家庭中,可通过 产前诊断,降低DMD的发生风险。如果能应用基因诊断方法,对所怀男胎排除DMD基因的突变,仍可生育正常男胎。








