- 神经影像: 视神经脊髓炎谱系疾病的MRI有哪些特点?
- 发布于 2015-08-12 07:30 来源:陈伟民医生
NMO是一种以严重的视神经炎和长节段横贯性脊髓炎(LETM)发作为特征的中枢神经系统炎症性疾病。过去的几十年里,我们对NMO的认识已取得巨大的进步。这主要得益于疾病特异性的自身抗体,NMO-IgG,以及随后其主要的自身靶抗原,水通道蛋白4(AQP-4)抗体的发现,从而使得NMO与MS区别开来,成为一个独立的疾病。
目前的诊断标准(指2006年的诊断标准)中,NMO的诊断依然需要出现视神经炎和脊髓炎。不过,AQP-4抗体的发现使得我们认识到除了经典的NMO外,还存在一组更为多样的临床表型,即所谓的“视神经脊髓炎谱系疾病(NMOSD)”。NMOSD不仅包含了血清AQP4抗体阳性的伴有特定脑部异常的限制型NMO(limited forms of NMO)或经典型NMO,还包括伴有其他自身免疫性疾病(例如SLE,干燥综合征等)的血清AQP4抗体阳性的患者。这种情况下,MRI在NMOSD与其他中枢神经系统炎症性疾病尤其是MS的鉴别中扮演着越来越重要的角色。因为治疗各异,因此将上述疾病区分开来很关键。此外,当前不断发展的MRI技术可帮助发现更多的特异性标志物,有助于阐明NMOSD潜在的致病机制。
NMO患者的早期头颅MRI研究发现了一些无法解释的临床无症状的、非特异性的白质病变。AQP4-IgG检测的出现更新了我们的认识,实际上很大一部分NMOSD患者存在脑部MRI的异常,常位于AQP4高表达区域。但AQP4表达不高的区域也可有脑部异常。虽然T2WI/FLAIR上非特异性的小点状或斑片状高信号是NMOSD最常见的影像学表现,但NMOSD某些病灶仍具有部位或形态学上的特征。
在AQP4抗体发现前,报道的脑部MR异常仅见于13-46%的NMO患者。但是,当不考虑头颅磁共振标准时,头颅MRI异常可达50-85%(参考修订版2006年NMO诊断标准)和51-89%(NMOSD血清抗体阳性患者)。此外,据报道,NMOSD发病初期即有头颅MRI异常的患者约43-70%。不同文献报道的头颅MRI异常的比例差异很大,一种可能解释是,随着疾病进展,头颅MRI异常出现的概率越来越高。在一组包含88例血清抗体阳性的儿童报道中,行头颅磁共振检查的患儿中有68%存在头颅异常,其中病灶在第三脑室周围区域(间脑)、第四脑室周围区域(脑干)、幕上和幕下的白质、中脑和小脑等部位更为明显。这与观测到45-55%的NMOSD患儿有发作性脑部症状这一现象是吻合的,临床表现包括眼肌麻痹、顽固性呕吐和呃逆、意识状态改变、严重的行为学改变、嗜睡、共济失调和癫痫发作。
NMOSD的头颅MRI表现分类
1、环绕脑室系统的室管膜周病变
(1)环绕第三脑室和导水管的间脑病变:环绕第三脑室和导水管的间脑病变部位包括丘脑、下丘脑、中脑前界,在NMOSD中已有报道(图1A)。这些病变通常是无症状的,但是有些患者可能会出现抗利尿激素分泌异常、嗜睡发作、低体温、低血压、睡眠过度、肥胖、甲状腺功能减退、高泌乳素血症、继发性闭经、溢乳和行为改变等症状。
(2)邻近四脑室的脑干背侧病变:邻近四脑室的脑干背侧包括极后区(area postrema)和孤束核的病变是NMOSD患者最为特异性的头颅MRI表现之一。见于7-46%的NMOSD患者,并且与顽固性呃逆、恶心、呕吐有高度相关性。这一区域为呕吐反射的中枢,有着较为疏松的血脑屏障,因而更容易受到AQP4-IgG的攻击。MRI和临床证据均提示极后区是NMOSD易受攻击的重要区域,后续研究发现该区域是血液循环中的IgG进入中枢神经系统的重要通道。40%的NMO患者这一区域有病理改变,但是没有明确的神经元、轴索或髓鞘缺失。延髓病变常与颈髓病变相连续,多呈线形(图1B.b)。这些病灶常与疾病的第一症状或者预示急性恶化有关。脑干病变可出现各种不同的临床表现,例如眼球震颤、构音障碍、吞咽困难、共济失调、眼肌麻痹等。
(3)环绕侧脑室的室管膜周病变:胼胝体病变见于12-40%的NMOSD患者。因为NMO和MS患者都常出现胼胝体病变,所以这一部位并不能作为NMO和MS的特异鉴别点。但是,MS的胼胝体病变常常是不连续的、卵圆形、垂直于侧脑室,多累及胼胝体下部(图2A)。而NMOSD病变位置与侧脑室很接近,紧贴室管膜内层(图1C.a)。NMOSD急性期的胼胝体病变常有明显水肿,且为多形性,形成“大理石样图案(marbled pattern)”,有时累及胼胝体压部全层,出现独特的“拱桥形图案(arch bridge pattern)”(图1C.b和C.c)。有时,胼胝体病变延续至大脑半球,形成广泛、融合的白质病变。在NMOSD的慢性期,胼胝体病变可逐渐缩小、信号减弱,甚至可以消失;但是,囊变和胼胝体萎缩都曾有过报道。某些临床表现,如认知功能和运动协调能力障碍等,可能与胼胝体受损有关,但证据并不是很充分。
2、大脑半球的白质病变
广泛、融合的大脑半球白质病变常呈瘤样(最大半径可以>3cm),或者沿白质纤维走行呈长纺锤状或放射状(图1D)。通常无占位效应。ADC上病灶弥散系数增高,提示可能为急性炎症相关的血管源性水肿(图1D.c),可能与可逆性后部脑病综合征(PRES)或Balo病混淆。相较于AQP4抗体阴性的患者,这些广泛的病变在AQP4抗体阳性的患者中更常见。疾病慢性期,这些大病灶趋向于缩小甚至消失,但有一些患者可出现囊样或空洞型改变。上述病变根据其累及区域的不同可引起各种症状,如偏瘫、脑病、视野缺损等。大片融合的大脑半球白质病变在NMOSD患儿中比较常见。伴随着灶周水肿和不同程度占位效应的肿瘤样病灶可类似急性播散性脑脊髓炎(ADEM)或中枢神经系统恶性肿瘤。
3、病变累及皮质脊髓束皮质
脊髓束受累可为单侧或双侧,病灶可能从大脑半球深部的白质通过内囊后肢延伸至中脑的大脑脚或者脑桥(图1E)。这些病灶连续,常为长节段,沿锥体束分布(图1E.c)。一些关于NMOSD患者的队列研究报道,约23-44%的患者存在皮质脊髓束的病灶,在其他研究中也偶有发现。有意思的是,和脑室周围不同,皮质脊髓束并不是AQP4高表达的区域,因此目前尚不清楚NMOSD患者这一区域常累及的原因。
4、非特异性病灶
T2WI/FLAIR上,皮质下区或深部白质区常可观察到非特异性的小点状(<3mm)或片状高信号,这是NMOSD最常见的头颅MRI异常(35-84%),通常无临床症状。
5、强化病灶
先前的一些研究报道9-36%的NMOSD患者头颅MRI可出现强化病灶,但具体比例尚不清楚。大多数病灶呈边界不清、模糊的多发片状强化,即所谓的“云雾状强化”(图1F.a)。这种云雾状的强化模式有助于与边界清楚的卵圆形或环状/开环状病灶(图2)鉴别,后者为MS较为典型的特征。侧脑室室管膜表面的线状强化(铅笔样病灶)也有报道(图1F.b)。边界清楚的结节样强化或脑膜强化也可见于NMOSD患者,但罕见。
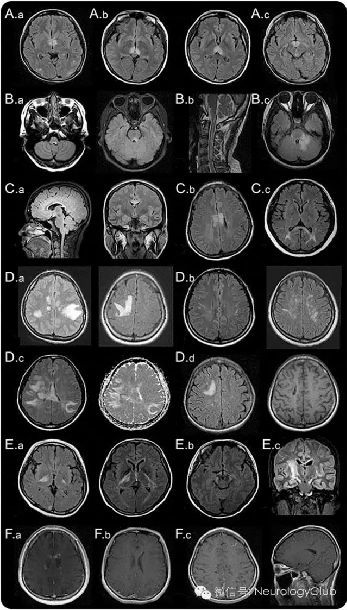
(Aa:环绕第三脑室和导水管的间脑病变;Ab:丘脑、下丘脑受累;Ac:中脑前界;Ba:邻近四脑室的脑干背侧病变;Bb:与颈髓病变相连续的延髓线状病变;Bc:肿胀弥漫的脑干背侧病变,累及小脑脚;Ca:邻近侧脑室、紧贴室管膜内层的胼胝体病变;Cb:胼胝体病变呈“大理石样图案” ;Cc:胼胝体病变呈“拱桥形图案”;Da:肿瘤样大脑半球白质病变;Db:长纺锤状病灶;Dc:ADC上病灶弥散系数增高,提示血管源性水肿;Dd:慢性期大脑半球病灶囊性变;Ea:内囊后肢皮质脊髓束病变;Eb:中脑大脑脚病变;Ec:沿锥体束的长节段病变;Fa:云雾状强化;Fb:侧脑室室管膜表面线状强化;Fc:脑膜强化)
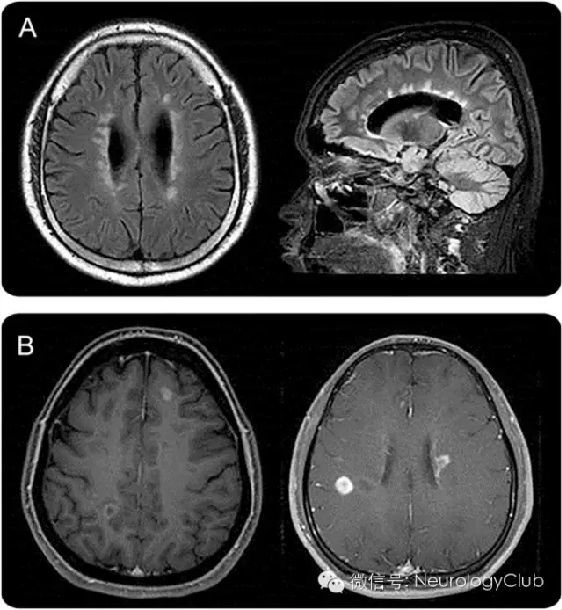
(A:侧脑室与胼胝体可见不连续的,卵圆形的,垂直于侧脑室的病灶;B:病灶呈卵圆形或开环强化,边界清楚)
NMOSD的视神经MRI表现
目前已有报道认为,在急性视神经炎期,T2WI和T1WI增强序列可见非特异性的视神经鞘增厚和视神经高信号。但因为MS患者视神经炎也可有类似改变,所以这一特点无法作为NMOSD的诊断支持点。现阶段的研究主要着眼于MS和NMOSD中视神经损伤磁共振的不同特点。NMOSD患者中,病变多累及包括视交叉在内的视神经后部,常同时有双侧视神经受累。因此,在临床实践中,当出现视神经长节段炎症,特别是同时存在双侧受累、病变向后延伸累及视交叉时,应考虑NMOSD的可能。
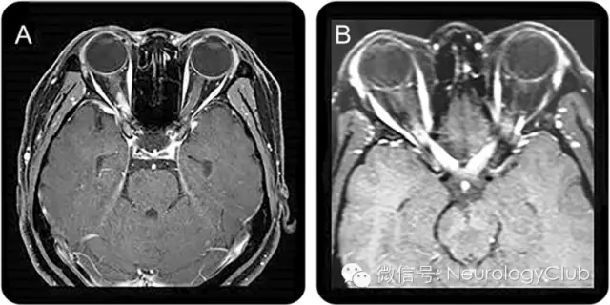
(A:右侧视神经后部强化病灶;B:双侧视神经后部/视交叉弥漫强化病灶)





